News from The Sosnick Group at The University of Chicago
Paper Published 09/22/2020

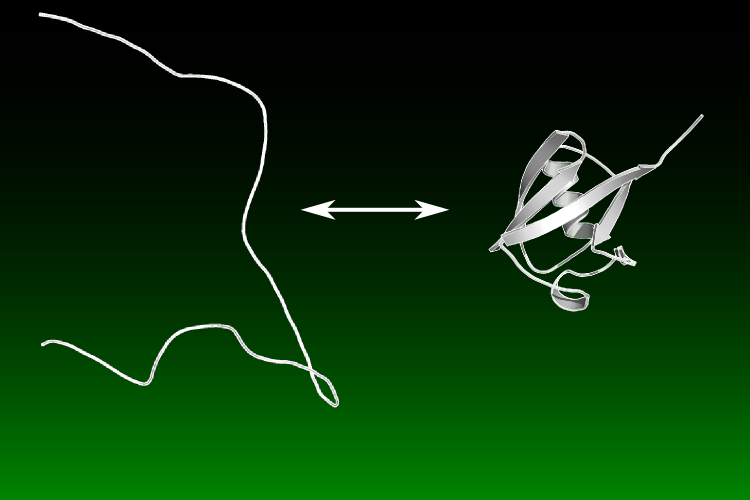
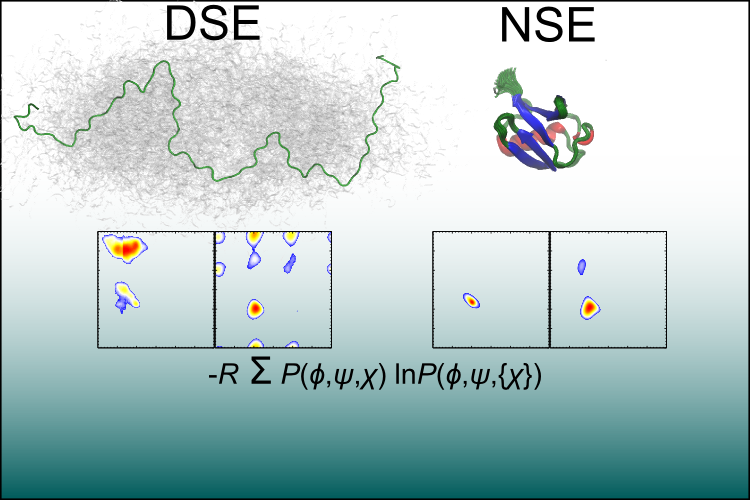
Congratulations to Joshua Riback and our collaborators in the Clark lab on having their recent paper, "Properties of protein unfolded states suggest broad selection for expanded conformational ensembles," published in PNAS!
Congratulations 12/03/2019

Congratulations to Kourtney Kroll on winning the David Grier Prize in Innovation in Biophysical Sciences for her project, "Engineering a Light Driven Molecular Motor", in collaboration with the Rock group.
Paper Published 10/30/2019


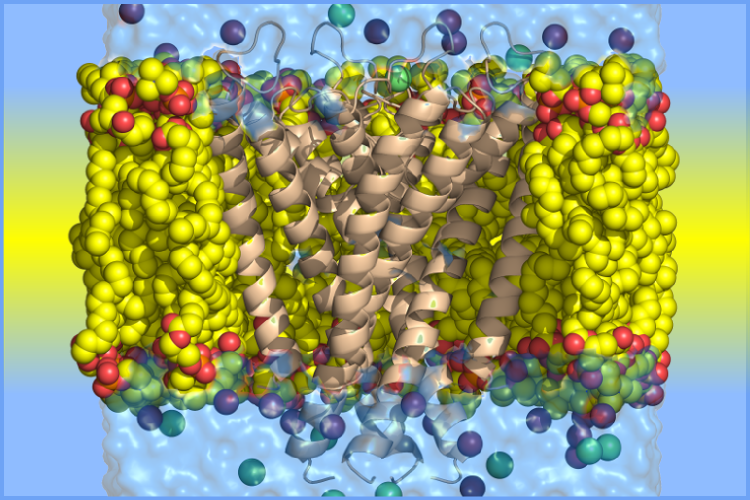
Congratulations to Zongan Wang and John Jumper on having their recent paper published, "On the Interpretation of Force-Induced Unfolding Studies of Membrane Proteins Using Fast Simulations", published in BIophysical Journal!
Welcome 07/17/2019
Welcome to Kourtney Kroll, who is joining our group as a new graduate student