Predicting Folding Dynamics
Predicting Folding Dynamics
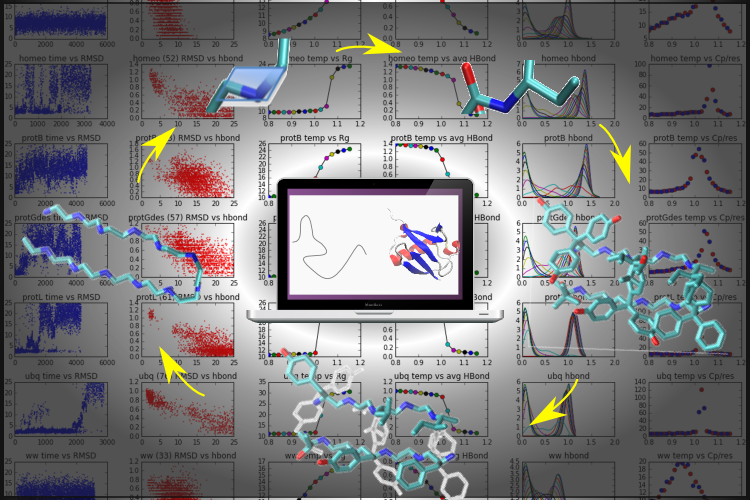
The traditional tradeoff in simulations between accuracy and speed is predicated on the assumption that force fields are typically well-parameterized. We re-examine this trade-off and show that a properly formulated coarse-grain model trained using machine learning methods can rival all-atom models for de novo protein folding and dynamics simulations. We use the contrastive divergence technique to train the force field directly from short simulation trajectories of 450 proteins beginning near their native structures. This method is applied to our recently developed Upside model, where the free energy for side chains are rapidly calculated at every time-step, allowing for a smooth energy landscape without steric rattling of the side chains (1). After this contrastive divergence training, the model is able to fold proteins up to ~100 residues de novo on a single core in CPU core-days (2).
The Upside model's success argues that simpler models that can be globally parameterized can rival more detailed but slower models whose parameterization is more challenging - more complexity does not necessarily equate to higher accuracy! Upside’s ready generation of Boltzmann ensembles allows for a wide range of studies of protein folding, dynamics and binding. Additionally, in studies that incorporate experimental or bioinformatics data, including contact predictions, Upside provides an inexpensive Bayesian prior distribution that may be updated using experimental information.
1. Accurate calculation of side chain packing and free energy with applications to protein molecular dynamics. https://doi.org/10.1371/journal.pcbi.1006342
2. Trajectory-based training enables protein simulations with accurate folding and Boltzmann ensembles in cpu-hours. https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006578
Approach
- Upside utilizes a number of unique features, including rapid side chain packing that enables MD simulations with only 3 backbone atoms whilre retaining considerable detail and avoiding sidechain "rattling", which greatly slows standard all-atom methods.
- We will continue to improve Upside and implement enhanced sampling methods to improve our accuracy and range of protein sizes
Related Posters
Extending Upside from protein folding to protein-protein interactions
