Membrane Proteins
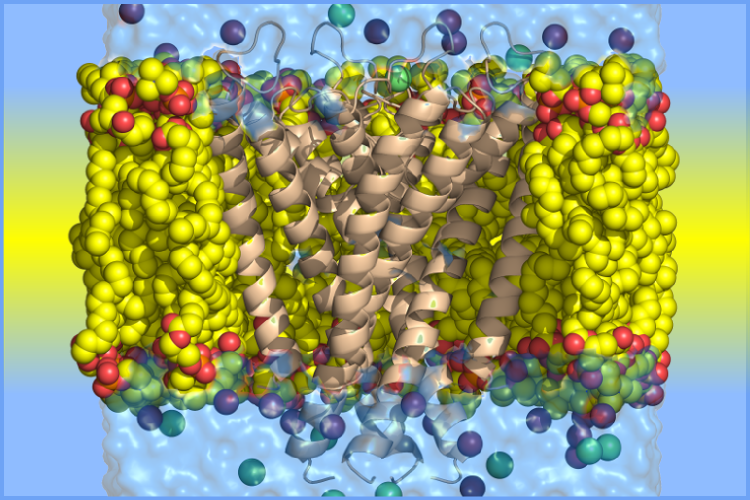
Membrane Proteins
Membrane protein folding does not necessarily follow the same empirical principles which describe soluble globular protein folding and dynamics. We are interested in extending the methods we've developed to membrane proteins.
For many potassium channels, tetramerization domains in either the N- or C-terminal of the pore domain assist in tetramer assembly by increasing local concentrations of monomeric subunits. The question we would like to understand is - to what extent do the pore domain monomers adopt a native-like structure found in tetramers?
Potassium Channel Monomers
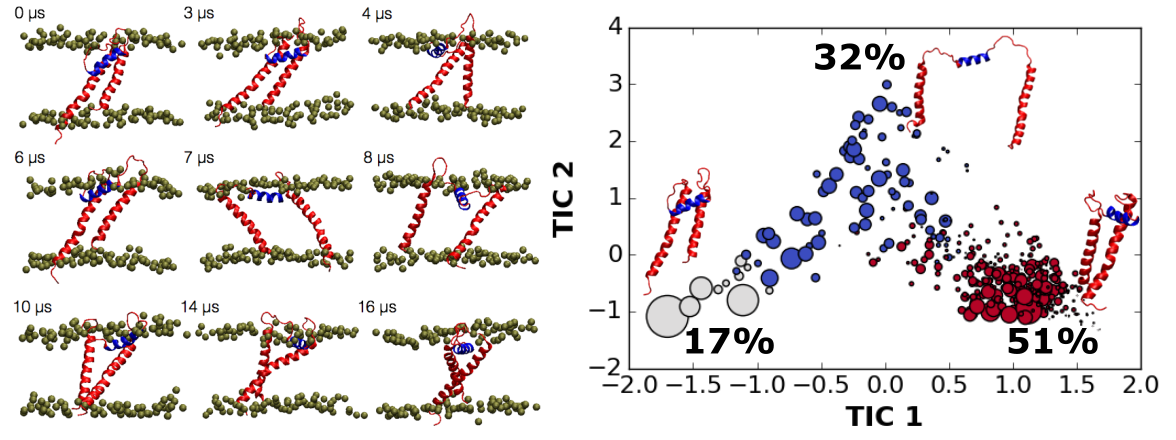
Potassium channels form a pore by juxtaposing four structurally identical or similar subunits around a central axis. The tetramerization process is thought to be assisted by intracellular domains at either N- or C-terminal of the pore domain by increasing subunit local concentrations, even though, the pore domain alone can still form functional tetramers. The question we are interested in addressing is what are the dynamics of potassium channel monomers before tetramerization. Using a combination of molecular dynamics (MD) simulations, Markov State Modeling (MSM) and Nuclear Magnetic Resonance (NMR) spectroscopy, we find that the potassium channel pore domain monomers in its lipid bilayer-like environment retain their nativelike secondary structures; however, each helix is highly dynamic respect to each other. With MD simulations and MSM, we find that the Kv1.2 pore domain monomer has 3 major states, and by trapping one of the major states using disulfide linkage, we are able to get the first glimpse of potassium channel pore domain monomer in bicelles through NMR.