Protein Folding
Protein Folding
We are interested in characterizing the general principles that govern protein folding. Despite being a long studied problem, many questions remain unresolved, for example the physical folding events that dictate the pathway as well as a concise understanding of the origins of the cooperative protein folding.
Our studies are consistent with the principle of Sequential Stabilization - that protein folding is a series of smaller folding events described by the concurrent formation of secondary and tertiary structure, e.g. a turn of helix forms which buries hydrophobic surface area. This incremental accumulation of structure mostly produces native- or unfolded-like regions during folding, as suggested by the frequent observation of ψ values of 0 or 1 as well as the cooperative pattern of protection factors for hydrogen exchange (HX).
"70% Rule of Folding"

We have characterized the transition state ensembles of several proteins and have found a common feature – that is the transition state achieves 70-80% of the native topology (as defined by relative contact order - Plaxco et al., 1998). This "70% Rule" supports the belief that topology is a critical feature of the transition state and supports the correlation between folding rate and topology. Furthermore, the high threshold places a strong constraint on feasible transition state structures for a particular native topology.
What makes a protein cooperative?

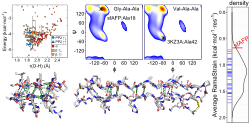
Our current views on the origin of folding cooperativity and stability are challenged by our studies of snowflea anti-freeze protein (sfAFP), which has a unique fold composed of 6 polyproline 2 (PP2) helices arranged in a 3x2 stack held together by inter-helical H-bonds. Despite lacking a hydrophobic core and being 46% Glycine, sfAFP is stable and folds in a two-state manner. sfAFP highlights that while we have a good, at least qualitative understanding of folding for typical, relatively small systems, much remains to be determined, especially the proper balance of the various forces that drive folding.